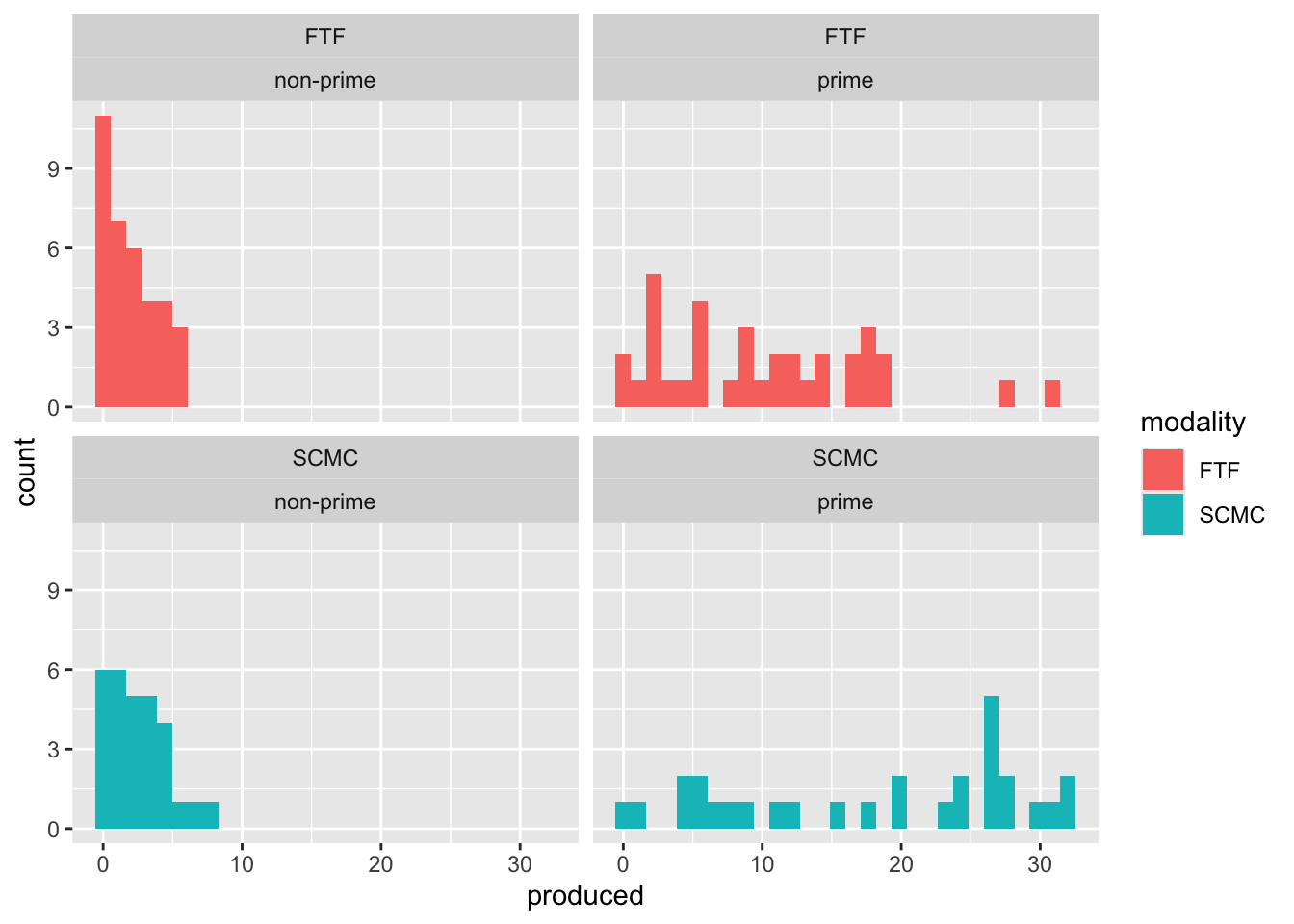
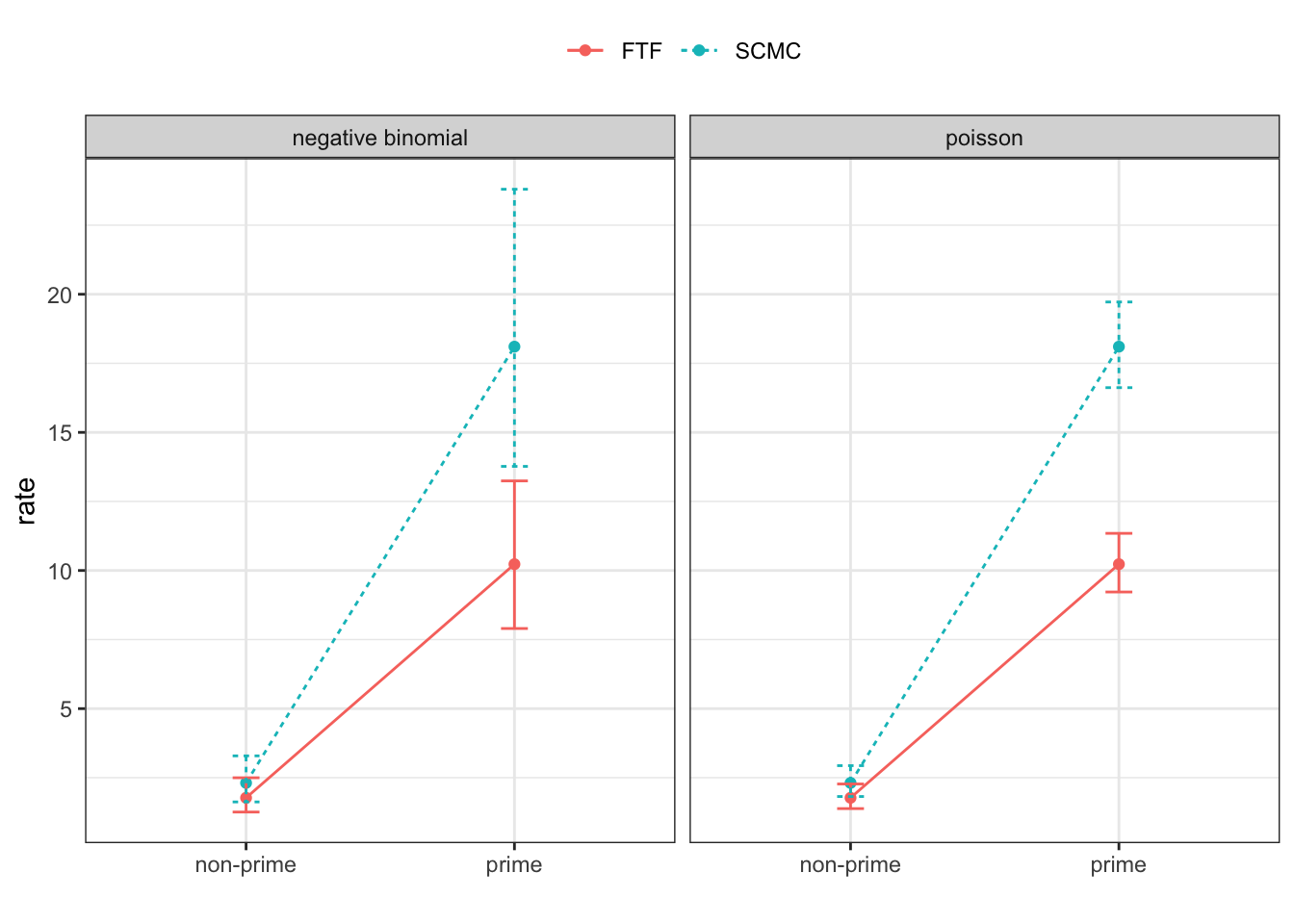
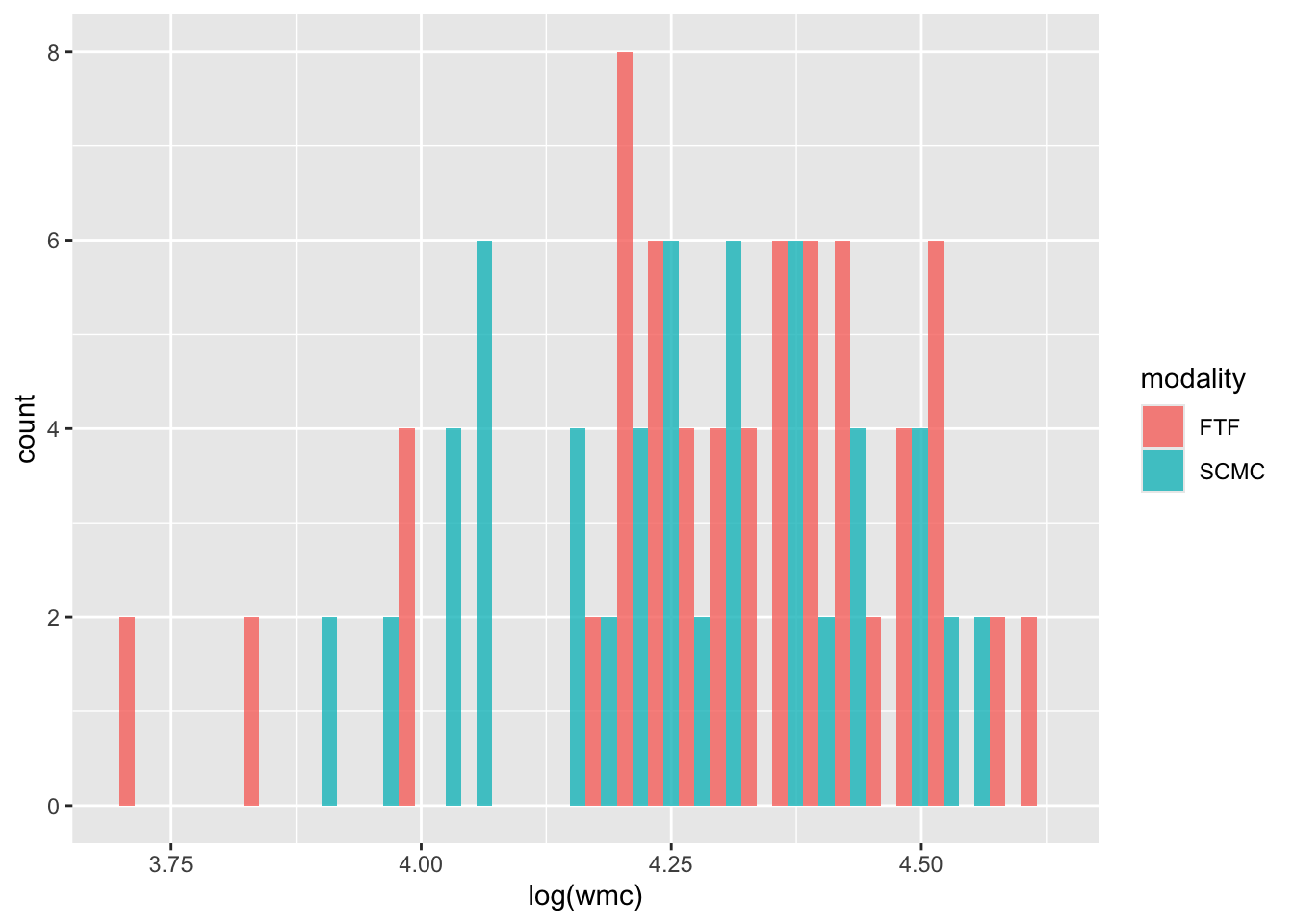
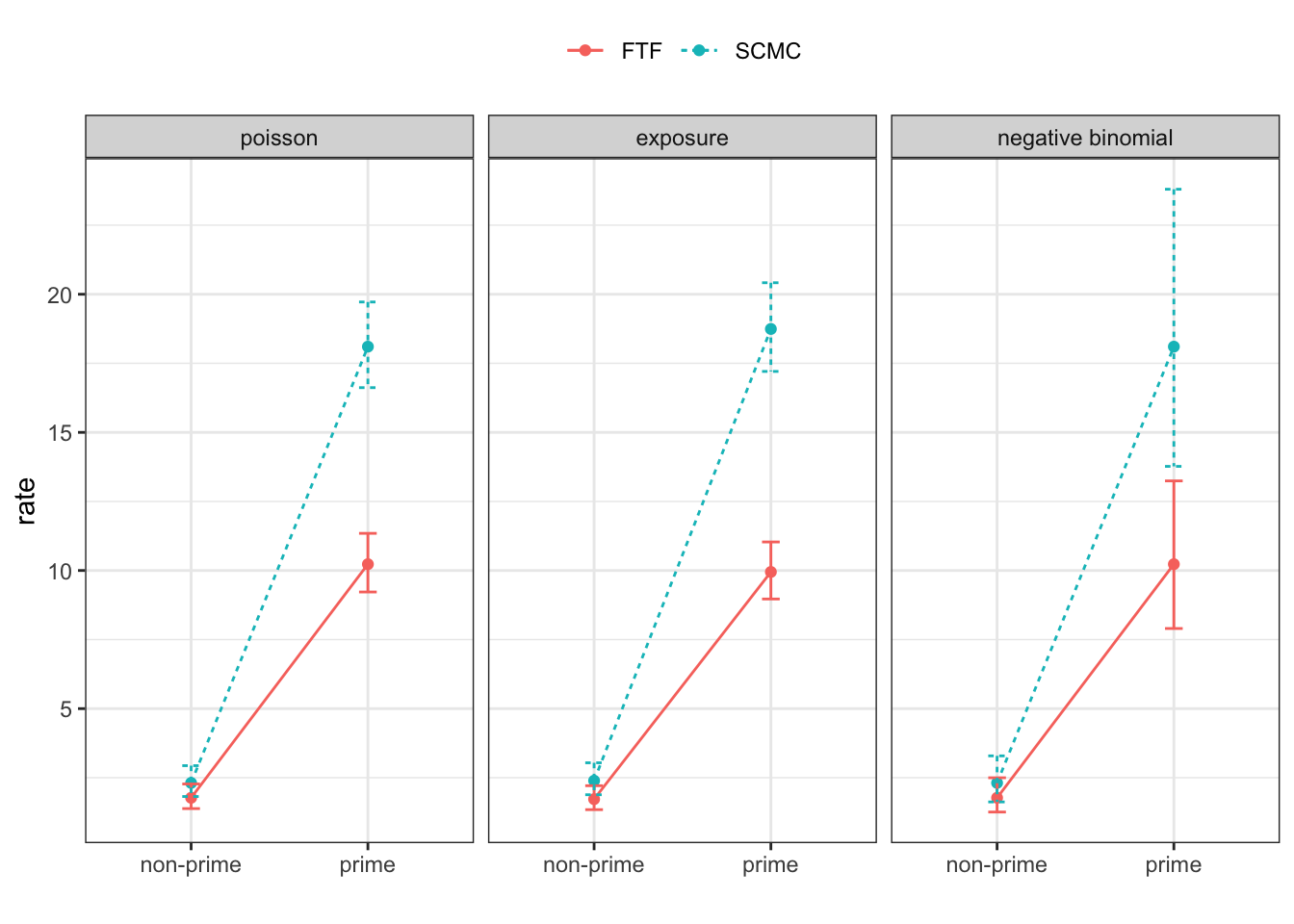
Rows: 4608 Columns: 12
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (5): verb, modality, test, session, trial_type
dbl (7): subject, score, trial_order, cloze, wmc, prod_pre, rec_pre
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
`summarise()` has grouped output by 'subject', 'modality', 'trial_type', 'prod_pre', 'rec_pre'. You can override using the `.groups` argument.
Rows: 128
Columns: 7
Groups: subject, modality, trial_type, prod_pre, rec_pre [128]
$ subject <dbl> 1, 1, 2, 2, 4, 4, 5, 5, 6, 6, 7, 7, 8, 8, 9, 9, 10, 10, 11,…
$ modality <chr> "FTF", "FTF", "FTF", "FTF", "FTF", "FTF", "FTF", "FTF", "FT…
$ trial_type <chr> "non-prime", "prime", "non-prime", "prime", "non-prime", "p…
$ prod_pre <dbl> 0, 0, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 0, 0, 3, 3, 2, 2,…
$ rec_pre <dbl> 6, 6, 5, 5, 5, 5, 6, 6, 14, 14, 7, 7, 4, 4, 0, 0, 6, 6, 6, …
$ wmc <dbl> 74, 74, 71, 71, 87, 87, 46, 46, 68, 68, 55, 55, 92, 92, 81,…
$ produced <dbl> 0, 2, 4, 19, 2, 2, 1, 11, 5, 18, 2, 10, 5, 5, 0, 0, 3, 6, 0…